BLAST analysis is simply defined as a sequencing tool that shows the sequence and relationship between different species using different search tools. BLAST stands for basic local alignment search tool. From this analysis we can easily analyse the sequence match with which type of organism with the help of similarities index.
A software and technique for comparing primary biological sequence data, such as the amino acid sequences of proteins or the nucleotides of DNA and/or RNA sequences, are known as BLAST (basic local alignment search tool) in the field of bioinformatics. Using a BLAST search, a researcher can find database sequences that resemble the alphabet over a predetermined threshold by comparing a subject protein or nucleotide sequence (referred to as a query) with a library or database of sequences.
BLAST analysis
BLAST, called the “Google of biological research” by The New York Times, is one of the most used bioinformatics tools for sequence searching. In bioinformatics study, it deals with a basic issue. Compared to alternative methods, such computing an ideal alignment, the heuristic technique it employs is far quicker. Although following algorithms may be even quicker, the emphasis on speed is essential to making the method work on the enormous genomic databases that are now accessible.
Basic Principle of Blast analysis
In addition to assisting in the identification of gene family members, BLAST may be used to infer functional and evolutionary links between sequences.
How to run BLAST software?
To run blast, its really simple. The following steps we have to follow while running the blast software:
- First of all, we have to go google and simple search NCBI blast.
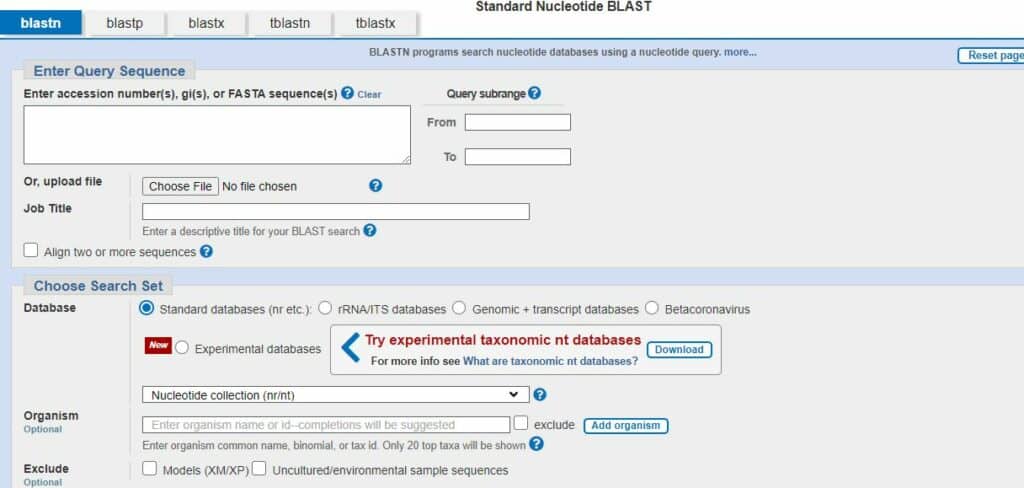
- The search tab is opened and simply, its depend upon us which type of sequencing we are running, just click that.
- For example if we are looking for nucleotide, simply click on nucleotide blast
- Then, the dialog box appear and we can simply enter accession number, fasta sequences, and simply go to the end and click on blast.
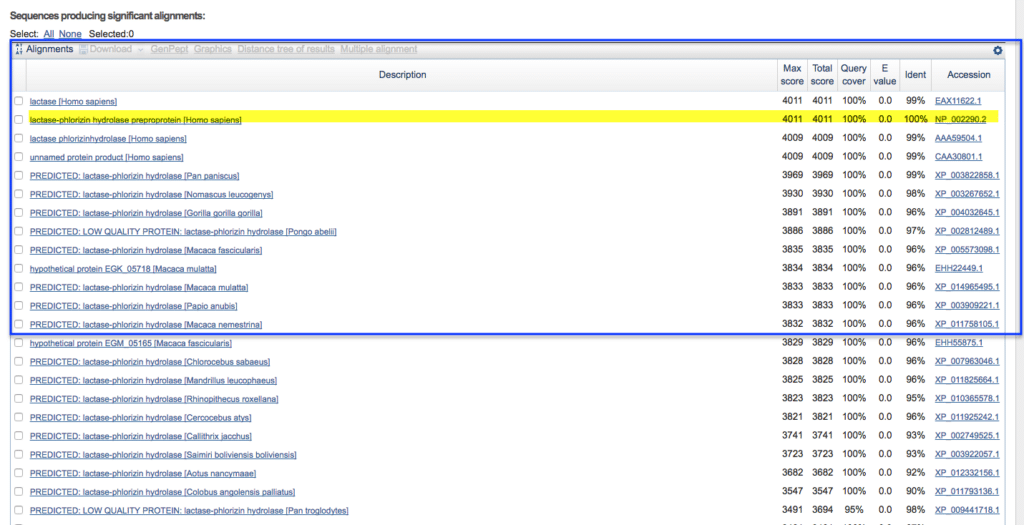
- The blast result will be seen within few seconds.
- Whatever our unknown sequences of bacteria, fungi will be showing similarity index matching with data bases but if the sequencing is not match with data bases then, it can be a new species.
Basic terms used in BLAST
After running blast, along with similarity index some terms are used:
- Similarity index
- Max score
- highest alignment score determined by adding the incentives for matching nucleotides or amino acids and the penalties for mismatches and gaps.
- Accession number
- A combination of letters and numbers, such as one letter followed by five digits (U12345) or two letters followed by six digits (AF123456), is typically used as an accession number that applies to the whole record. In accordance with the kind of sequence record, certain accessions could be lengthier.
- Total score
- the total alignment scores—calculated across all segments—of all segments from the same database sequence that match the query sequence.
- Query cover
- The amount of the query sequence (your specimen) that overlaps the reference sequence is known as the query cover. Typically, BLAST results do not try to match a whole sequence. For the initial triage, a high Query Cover value is in the 70%+ range.
- Accession length
- the number of nucleotides or amino acids in the accession number-identified result sequence.
Similarity %
A high Identity value for the initial triage often corresponds to a sequence similarity of 98–100%. The proportion of base pairs that are identical between the sequence of your specimen and the reference specimen is reported by the identity.
Types of BLAST
Traditionally there are five types of BLAST:
- BLASTN
- BLASTP
- BLASTX
- TBLASTN
- TBLASTX
- BLASTN
- Nucleotides
- Annotating genomic DNA, grouping sequencing reads, screening for repetitive elements, mapping oligonucleotides, cDNAs, and PCR products to a genome, and vector clipping are some examples.
- Nucleotides
- BLASTP
- Protein
- gathering similar proteins for phylogenetic analysis and locating shared areas in proteins.
- Protein
- BLASTX
- nucleotides translated to protein
- Identifying the genomic DNA that codes for proteins; figuring out whether a cDNA represents a recognized protein.
- nucleotides translated to protein
- TBLASTN
- Protein
- locating transcripts that, presumably, come from different species and that resemble a certain protein; mapping a protein to genomic DNA.
- Protein
- TBLASTX
- Nucleotide translated to protein
- Genome or transcript level cross-species gene prediction; looking for genes overlooked by conventional approaches or not yet in protein databases.
- Nucleotide translated to protein
Importance of Blast
- By comparing local similarities between protein or nucleotide sequences, the Basic Local Alignment Search Tool (BLAST) may identify these areas. The program determines the statistical significance of matches by comparing nucleotide or protein sequences to sequences in a database.
- There are various uses for BLAST. These include detecting domains, creating phylogenies, identifying species, mapping DNA, and comparing. Using BLAST, you might be able to accurately identify a species or discover closely related species.
Reference
https://etutorials.org/Misc/blast/Part+III+Practice/Chapter+5.+BLAST/5.1+The+Five+BLAST+Programs/